





Акрамя тэхналогій, сінтэз гліказідаў заўсёды цікавіў навуку, бо гэта вельмі распаўсюджаная рэакцыя ў прыродзе. Нядаўнія працы Шміта, Тошымы і Тацуты, а таксама шматлікія цытаваныя ў іх спасылкі апісваюць шырокі спектр сінтэтычных магчымасцей.
Пры сінтэзе гліказідаў шматцукровыя кампаненты спалучаюцца з нуклеафіламі, такімі як спірты, вугляводы або бялкі. Калі патрабуецца селектыўная рэакцыя з адной з гідраксільных груп вуглявода, усе астатнія функцыі павінны быць абаронены на першым этапе. У прынцыпе, ферментатыўныя або мікробныя працэсы, дзякуючы сваёй селектыўнасці, могуць замяніць складаныя этапы хімічнай абароны і зняцця абароны для селектыўнага выдалення гліказідаў у пэўных рэгіёнах. Аднак з-за доўгай гісторыі алкілгліказідаў прымяненне ферментаў у сінтэзе гліказідаў не атрымала шырокага вывучэння і ўжывання.
З-за магчымасці выкарыстання адпаведных ферментных сістэм і высокіх выдаткаў на вытворчасць, ферментатыўны сінтэз алкілполігліказідаў не гатовы да пераходу на прамысловы ўзровень, і перавага аддаецца хімічным метадам.
У 1870 годзе М.А. Колі паведаміў пра сінтэз «ацэтахларгідрозы» (1, малюнак 2) шляхам рэакцыі дэкстрозы (глюкозы) з ацэтылхларыдам, што ў рэшце рэшт прывяло да гісторыі шляхоў сінтэзу гліказідаў.
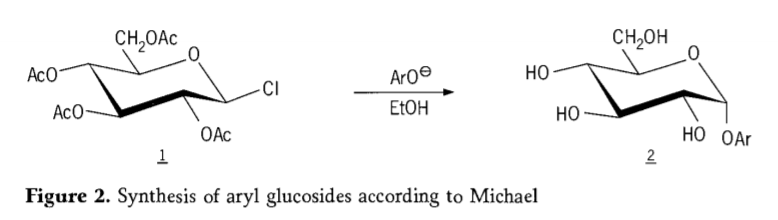
Тэтра-0-ацэтыл-глюкапіраназілгалагіны (ацэтагалагаглюкозы) пазней аказаліся карыснымі прамежкавымі прадуктамі для стэрэаселектыўнага сінтэзу чыстых алкілглюказідаў. У 1879 годзе Артур Майкл паспяхова атрымаў пэўныя, крышталізуемыя арылгліказіды з прамежкавых прадуктаў і феналятаў Колі (Аро-, малюнак 2).
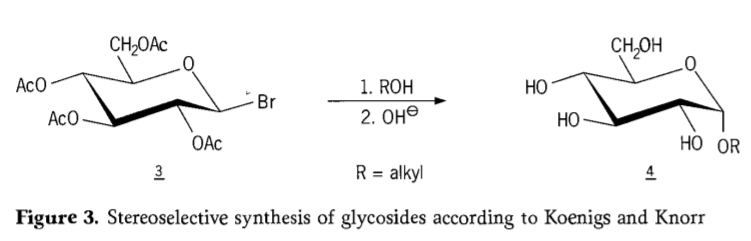
У 1901 годзе Майкл правёў сінтэз шырокага спектру вугляводаў і гідраксільных агліконаў, калі У. Кёнігс і Э. Кнор прадставілі свой палепшаны стэрэаселектыўны працэс гліказідавання (малюнак 3). Рэакцыя ўключае замяшчэнне SN2 на анамерным вугляродзе і працякае стэрэаселектыўна з інверсіяй канфігурацыі, утвараючы, напрыклад, α-глюказід 4 з β-анамера прамежкавага прадукту ацэабромглюкозы 3. Сінтэз Кёнігса-Кнора адбываецца ў прысутнасці срэбных або ртутных прамотараў.
У 1893 годзе Эміль Фішэр прапанаваў прынцыпова іншы падыход да сінтэзу алкілглюказідаў. Гэты працэс цяпер добра вядомы як «гліказідаванне Фішэра» і ўяўляе сабой кіслотна-каталізаваную рэакцыю глікоз са спіртамі. Тым не менш, любы гістарычны справаздачу павінен таксама ўключаць першую апісаную ў 1874 годзе спробу А. Гот'е пераўтварыць дэкстрозу бязводным этанолам у прысутнасці салянай кіслаты. З-за памылковага элементнага аналізу Гот'е лічыў, што атрымаў «дыглюкозу». Пазней Фішэр паказаў, што «дыглюкоза» Гот'е насамрэч была ў асноўным этылглюказідам (малюнак 4).
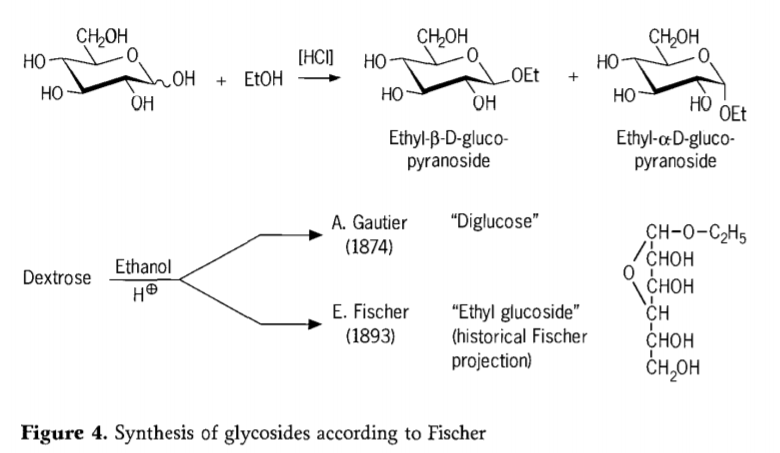
Фішэр правільна вызначыў структуру этылглюказіду, што відаць з прапанаванай гістарычнай фуранозідавай формулы. Фактычна, прадукты гліказідавання Фішэра — гэта складаныя, пераважна раўнаважныя сумесі α/β-анамераў і ізамераў піранозіду/фуранозіду, якія таксама ўтрымліваюць выпадкова звязаныя гліказідныя алігамеры.
Адпаведна, асобныя малекулярныя часціцы няпроста вылучыць з рэакцыйных сумесяў Фішэра, што ў мінулым было сур'ёзнай праблемай. Пасля некаторага ўдасканалення гэтага метаду сінтэзу Фішэр пазней выкарыстаў сінтэз Кёнігса-Кнора для сваіх даследаванняў. Выкарыстоўваючы гэты працэс, Э. Фішэр і Б. Хельферых упершыню паведамілі пра сінтэз доўгаланцуговага алкілглюказіду, які праяўляе павярхоўна-актыўныя ўласцівасці, у 1911 годзе.
Яшчэ ў 1893 годзе Фішэр правільна заўважыў важныя ўласцівасці алкілгліказідаў, такія як іх высокая ўстойлівасць да акіслення і гідролізу, асабліва ў моцна шчолачных асяроддзях. Абедзве характарыстыкі каштоўныя для алкілполігліказідаў у прымяненні ў якасці павярхоўна-актыўных рэчываў.
Даследаванні, звязаныя з рэакцыяй гліказідавання, усё яшчэ працягваюцца, і ў апошні час было распрацавана некалькі цікавых шляхоў атрымання гліказідаў. Некаторыя працэдуры сінтэзу гліказідаў коратка прадстаўлены на малюнку 5.
У цэлым, працэсы хімічнага гліказідавання можна падзяліць на працэсы, якія прыводзяць да складаных алігамерных раўнаважак пры кіслотна-каталізаваным гліказільным абмене.
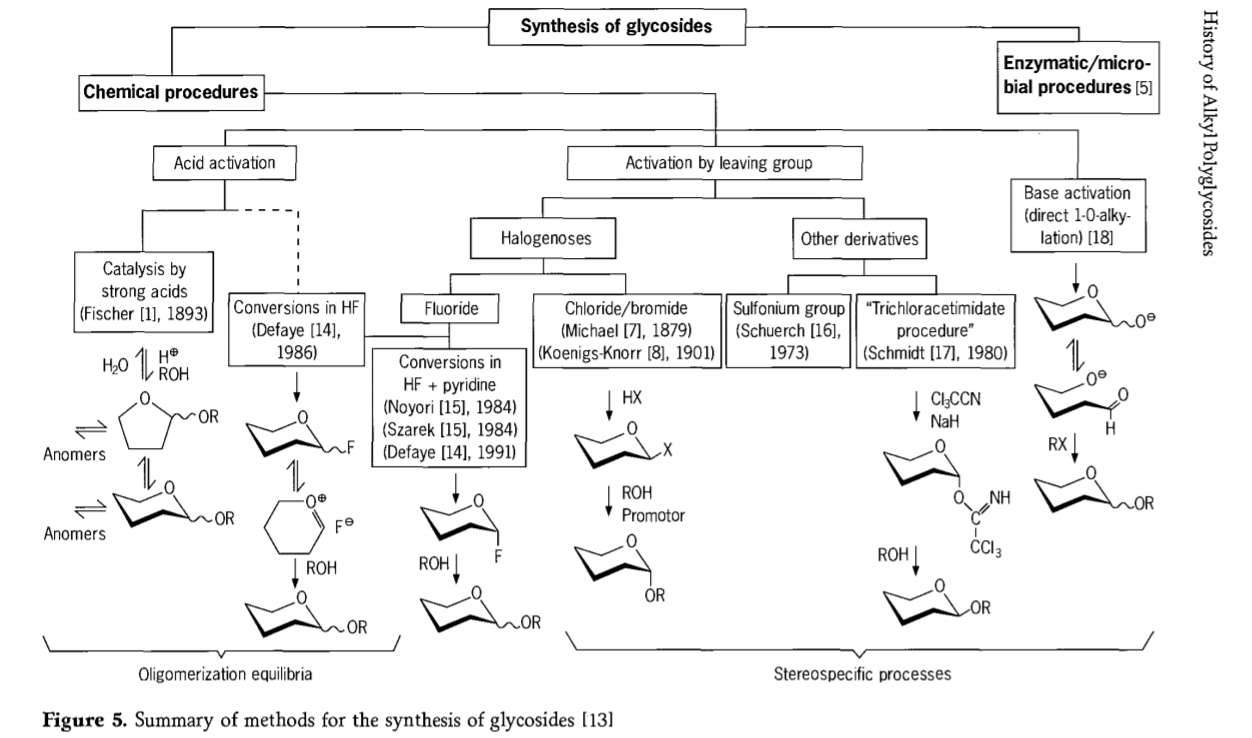
Рэакцыі на адпаведна актываваных вугляводных субстратах (гліказідныя рэакцыі Фішэра і рэакцыі фторнага вадароду (HF) з неабароненымі малекуламі вугляводаў) і кінетычна кантраляваныя, незваротныя і ў асноўным стэрэатаксічныя рэакцыі замяшчэння. Другі тып працэдуры можа прывесці да ўтварэння асобных відаў, а не ў складаных сумесях рэакцый, асабліва ў спалучэнні з метадамі захавання груп. Вугляводы могуць пакідаць групы на эктапічным вугляродзе, такія як атамы галагенаў, сульфанілы або трыхларацетымідатныя групы, або актывавацца асновамі перад пераўтварэннем у трыфлатныя эфіры.
У прыватным выпадку гліказідавання ў фторыстым вадародзе або ў сумесях фторыстага вадароду і пірыдзіну (пірыдыніевага полі[фторысты вадарод]), гліказілфтарыды ўтвараюцца in situ і плаўна пераўтвараюцца ў гліказіды, напрыклад, са спіртамі. Было паказана, што фторысты вадарод з'яўляецца моцна актывуючым, нераскладальным рэакцыйным асяроддзем; раўнаважная аўтакандэнсацыя (алігамерызацыя) назіраецца падобна працэсу Фішэра, хоць механізм рэакцыі, верагодна, адрозніваецца.
Хімічна чыстыя алкілгліказіды падыходзяць толькі для вельмі спецыяльных ужыванняў. Напрыклад, алкілгліказіды паспяхова выкарыстоўваюцца ў біяхімічных даследаваннях для крышталізацыі мембранных бялкоў, такіх як трохмерная крышталізацыя порыну і бактэрыярадапсіну ў прысутнасці актыл-β-D-глюкапіраназіду (далейшыя эксперыменты, заснаваныя на гэтай працы, прывялі да Нобелеўскай прэміі па хіміі для Дайзенхофера, Хубера і Мішэля ў 1988 годзе).
Падчас распрацоўкі алкілполігліказідаў стэрэаселектыўныя метады выкарыстоўваліся ў лабараторным маштабе для сінтэзу розных мадэльных рэчываў і вывучэння іх фізіка-хімічных уласцівасцей. З-за іх складанасці, нестабільнасці прамежкавых прадуктаў, а таксама колькасці і крытычнага характару адходаў працэсу, сінтэзы тыпу Кенігса-Кнора і іншыя метады ахоўных груп стваралі б значныя тэхнічныя і эканамічныя праблемы. Працэсы тыпу Фішэра параўнальна менш складаныя і лягчэй выконваць у камерцыйных маштабах і, адпаведна, з'яўляюцца пераважным метадам вытворчасці алкілполігліказідаў у вялікіх маштабах.
Час публікацыі: 12 верасня 2020 г.